
 Faculty of Science, Technology and Medicine
Faculty of Science, Technology and Medicine Faculty of Law, Economics and Finance
Faculty of Law, Economics and Finance Faculty of Humanities, Education and Social Sciences
Faculty of Humanities, Education and Social Sciences Interdisciplinary Centre for Security, Reliability and Trust
Interdisciplinary Centre for Security, Reliability and Trust Luxembourg Centre for Systems Biomedicine
Luxembourg Centre for Systems Biomedicine Luxembourg Centre for Contemporary and Digital History
Luxembourg Centre for Contemporary and Digital History Luxembourg Centre for European Law
Luxembourg Centre for European Law Luxembourg Centre for Socio-Environmental Systems
Luxembourg Centre for Socio-Environmental SystemsOur research
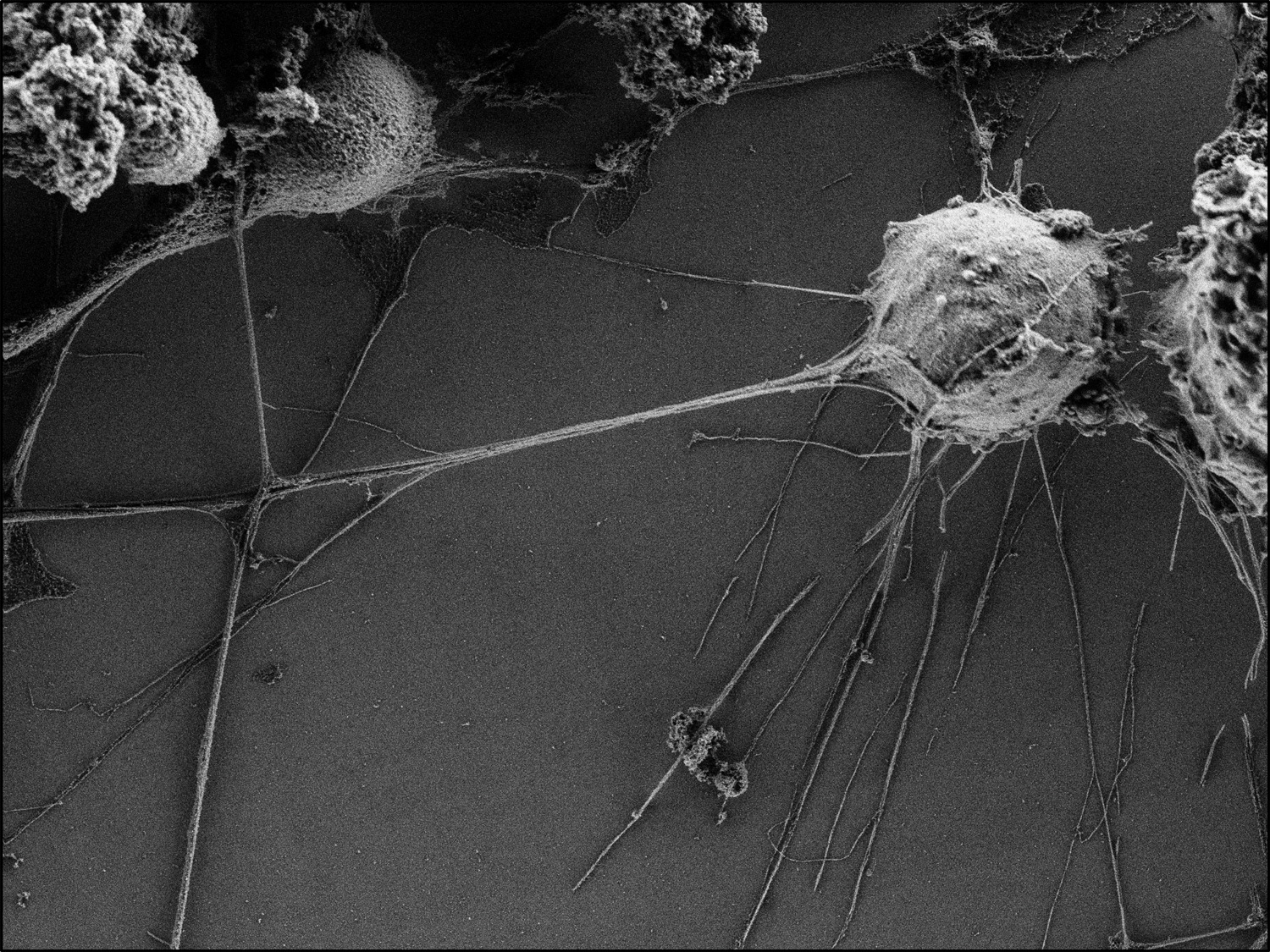
TNT-like structures between neurons and microglia, visualised by scanning electron microscopy
Neuroinflammation is a hallmark of neurodegenerative diseases such as Alzheimer’s (AD). AD is characterised by the accumulation of extracellular amyloid-beta (Aβ) in amyloid plaques and intracellular tau in neurofibrillary tangles (NFTs). The activation of the innate immune cells of the brain, called microglia, seems to be involved in the development and progression of this and other neurodegenerative diseases. To date, the underlying mechanisms in the transition of an initially protective to a chronic and harmful neuroinflammatory response are unknown.
The group aims at understanding molecular mechanisms of inflammatory regulation in a variety of neurodegenerative diseases, using novel mouse models and state-of-the-art techniques like two-photon imaging, transcriptome analysis and induced pluripotent stem cells (iPSCs). The group also studies the cellular interactions between microglia, neurons, astrocytes, and oligodendrocytes through tunnelling nanotubes. From a translational perspective, the goal of the group is to develop new biomarkers and insights into medical interventions around various aspects of neuroinflammation involved in neurodegenerative and cerebrovascular diseases.
Our research projects
This section introduces current projects of the Neuroinflammation group.
-
Duration:
2023-2028
-
Funding source:
FNR PEARL Programme / University of Luxembourg
-
Researchers:
Michael T. HENEKA, Sean-Patrick H. RIECHERS, Sofie-Helene SEIBEL, Pablo BOTELLA LUCENA, Dimitri BUDINGER, Vivian Song-Minkyung BAKER, Michele Fabrizio CALIANDRO, Hugo Filipe RANGEL DA COSTA, Beatriz ALMEIDA, Zhe LIU (past member), Mariana PIRES (past member)
-
Partners:
-
Description:
Dementia has been identified by the Word Health Organization as a major and global health issue. Indeed, we expect an increase from currently 55 million dementia cases to about 150 million in 2050. Approximately two thirds of all dementia patients are suffering from Alzheimer’s disease (AD), a neurodegenerative disorder that leads to memory dysfunction, behavioral disturbances, and loss of all higher cognitive functions.
PEARL MINIALZ aims to investigate direct cellular interactions between microglia and neurons by tunnelling nanotubes (TNTs). Initial data suggest that microglia can establish TNTs with neurons, enabling them to clear pathological protein aggregates and facilitate organelle trafficking between cells. The transport of aggregates via TNTs in AD and its degradation by microglia may play a critical role in preserving neuronal function and integrity. In this project, we will use different disease models to address this question. We will use microglia and neurons differentiated from patient-derived induced pluripotent stem cells (iPSC), as well as primary murine cells and cell lines, to comprehensively investigate, in the context of AD, the formation of TNTs and the functional transfer/modification of cargo through these structures using confocal live imaging techniques. We will further validate findings in fish and rodent models with state-of-the-art 3D super-resolution microscopy and multi-photon in vivo microscopy. Finally, we will validate our findings in microglia and neurons differentiated from iPSCs derived from patients.
In addition, we will also assess whether modulating TNT formation between neurons and microglia could open new therapeutic or diagnostic avenues for the treatment of AD.
-
Project details (PDF):
-
Duration:
2023-2027
-
Funding source:
EU Joint Programme – Neurodegenerative Disease Research (JPND)
-
Researchers:
Hilmi Jaufer THAMEEMUL ANSARI
-
Partners:
- Tarja Malm, University of Eastern Finland, Finland
- Meaghan O’Reilly, Sunnybrook Research Institute/University of Toronto, Canada
- Arn M.J.M. van den Maagdenberg, Leiden University Medical Center, Netherlands
- Denis Vivien, INSERM – Caen Normandie University and Hospital, France
- Signe Mežinska, University of Latvia, Latvia
- Else Tolner, Leiden University Medical Centre, Netherlands
-
Description:
The project aims to elucidate key cellular–molecular targets and mechanisms by which the mechanoreceptor PIEZO1 and other mechanoreceptors are used by microglia to sense Aβ deposit stiffness, and to clear Aβ deposits upon cellular activation. We will treat induced pluripotent stem cell (iPSC) differentiated microglia and rodent primary microglia with mechanoreceptor agonists, mechanical stimulation, and stimuli combinations to assess their role in microglial phagocytosis, lysosomal functions, and inflammatory processes in an AD context. We will use CRISPR-Cas9 constructs or antagonists against respective membrane proteins in iPSC-derived microglia to study loss of function phenotypes.
In murine models of cerebral amyloidosis, we will assess microglial morphology at Aβ deposits and their clearance by microglia using multi-photon in vivo laser scanning microscopy. Results will be validated by FACS-mediated isolation from the adult brain and analysis of microglia containing phagocytosed Aβ. Isolated microglia from brains will also undergo scRNA-Seq in order to dissect pathways involved in mechanoreceptor-dependent microglial immune activation. Furthermore, we will evaluate neuronal network activity and behavioral phenotypes after activating mechanoreceptors in the models described above. We will monitor neuronal calcium transients and study learning and memory behavior.
In summary, we will generate new information on possible therapeutic effects of microglial mechanoreceptor activation with the aim to maintain microglial homeostasis and clearance capacity.
-
Project details (PDF):
-
Duration:
2023 – 2025
-
Funding source:
European Molecular Biology Organisation (EMBO) Fellowship
-
Researchers:
Bora TASTAN
-
Partners:
-
Description:
Alzheimer’s disease (AD) is characterised by three hallmark pathologies – the accumulation of amyloid-beta (Aβ) in extracellular plaques, the aggregation of hyperphosphorylated tau in intracellular neurofibrillary tangles (NFT), and neuroinflammation. However, little is currently known about how these pathologies connect. Research over the past decade has shown that microglia, the innate immune cells of the brain, can sense aggregated Aβ. This leads to the assembly of the NLRP3 inflammasome, which consists of the intracellular pattern recognition receptor NLRP3, the adaptor protein ASC, and the effector enzyme caspase-1. Subsequently, microglia produce and secrete several inflammatory mediators, including IL-1β and ASC protein complexes termed as ASC specks. Interestingly, Aβ plaque formation seems to precede tau pathology, and numerous studies have demonstrated that Aβ fibrils can induce tau seeding and spreading. While microglia depletion, as well as genetic knockout of ASC, were found to reduce spread in an in vivo tau spread model, the influence of the NLRP3 inflammasome on neuronal tau pathology and tau pathology downstream of Aβ has yet to be elucidated. We hypothesize that genetic knockout of either ASC or Nlrp3 not only affects the transcriptome in microglia, but also induces transcriptomic changes in astrocytes and neurons in a non-cell-autonomous manner. We further predict that the NLRP3 inflammasome is important for cellular senescence in tauopathies and for the formation and maintenance of neuronal spines. We will also evaluate the ability of the NLRP3 inflammasome to modulate Aβ-induced tau pathology and investigate if isolated ASC specks are able to induce tau phosphorylation and tangle formation.
-
Project details (PDF):
-
Duration:
2022 – 2026
-
Funding source:
The Gerhard and Ilse Schick Foundation (Gerhard und Ilse Schick Stiftung)
-
Researchers:
Masanori ITAKURA, Beatriz ALMEIDA,Adrien RAUH (past member), Archontia Maria ANANIADOU (past member)
-
Partners:
AC Immune SA, Switzerland
-
Description:
In Alzheimer’s Disease (AD), evidence show that ASC speck formation downstream of the NLRP3 inflammasome activation underlies the deposition of amyloid-beta (Aβ). ASC specks can be defined as micrometer sized protein complexes with complex physiological roles arising from ASC-dependent inflammasome activation. Once released into the extracellular space in response to pyroptosis, ASC specks were shown to directly interact with Aβ and serve as a seed for Aβ aggregation in vitro and in vivo. ASC specks are therefore promising targets for novel therapeutic approaches aiming at slowing down AD progression. Our project consists of using diverse molecular biology and imaging techniques to further define the dynamics of pyroptosis and ASC speck release. We will further evaluate if anti-ASC antibodies prevent or slow down Aβ aggregation induced by ASC specks in vitro and in vivo.
-
Project details (PDF):
-
Duration:
2023 – 2025
-
Funding source:
University of Luxembourg (IAS Audacity)
-
Researchers:
Laurent MARICHAL, Archontia Maria ANANIADOU (past member)
-
Partners:
Theoretical Chemical Physics research group (Prof. Alexandre TKATCHENKO) at the University of Luxembourg
-
Description:
During inflammatory responses, reactive oxygen and nitrogen species are produced and interact promiscuously with endogenous molecules, including aggregation-prone peptides. In the context of amyloid diseases, such as Alzheimer’s disease (AD), it is well-established that inflammatory damage can exacerbate the aggregation and neurotoxicity of amyloid-beta (Aβ) peptides. However, the precise mechanisms underlying this process remain unclear. Notably, under nitro-oxidative stress Aβ can undergo a post-translational modification called nitration, as it was shown in Alzheimer’s disease (AD) patient brains. Furthermore, nitrated Aβ seems to form aggregates at a different rate compared to unmodified Aβ. The exact link between nitrated Aβ and AD is currently not understood. Therefore, the goal of this project is to combine experimental and computational approaches in order unravel the mechanisms behind the effect of Aβ nitration in the context of AD. This project involves a collaboration with the Theoretical Chemical Physics department at UL.
-
Project details (PDF):
-
Duration:
2023-2026
-
Funding source:
PI funding (University of Luxembourg)
-
Researchers:
Shilauni DADWAL
-
Partners:
-
Description:
The regulation of the NLRP3 inflammasome, a critical component of the innate immune system, is a complex and dynamic process. In this project, we will delve into how NEK7, MARK4, GBP5, PKR, AKT, BTK, and other proteins regulate the NLRP3 inflammasome, and explore their potential connections in the downstream cascade of ASC-dependent and independent pathways.
NEK7 has been identified as a critical player in NLRP3 inflammasome activation. Indeed, NEK7 binding induces a conformational change in NLRP3, allowing its oligomerization and subsequent inflammasome assembly. MARK4 kinase is also implicated in NLRP3 inflammasome regulation. Guanylate-binding proteins (GBPs) can promote NLRP3 inflammasome assembly and cytokine release during specific infections. Protein kinase PKR has been shown to negatively regulate NLRP3 inflammasome activation by phosphorylating and inhibiting RIPK1 kinase, which is involved in inflammasome signaling. The AKT kinase can phosphorylate NLRP3 and promote its degradation, thus acting as a direct negative regulator. Bruton’s tyrosine kinase (BTK) is associated with NLRP3 inflammasome activation in the context of autoimmune diseases, and BTK inhibitors have shown promise in reducing inflammasome-driven inflammation.
The interconnections between these inflammatory cascade regulators are an exciting area of research. Some of these kinases may act sequentially or synergistically, while others may counteract each other’s effects. Elucidating these interactions will be critical to understand the precise regulatory mechanisms of NLRP3 inflammasome activation in microglia, and consequently develop targeted therapies for inflammatory diseases. Thus, this project aims to provide a comprehensive understanding of how the NLRP3 inflammasome is regulated in canonical, non-canonical, and alternative pathways in rodent and human microglia. By uncovering the intricate interplay between these regulators, we can contribute to a more nuanced understanding of inflammasome biology, which has significant implications for therapeutic interventions in inflammatory diseases.
-
Project details (PDF):
-
Duration:
2023-2027
-
Funding source:
NextImmune2 Doctoral Training Unit (FNR PRIDE Programme)
-
Researchers:
Shreya Yashraj REGE
-
Partners:
-
Description:
Activation of microglia and subsequent formation of NACHT, LRR and PYD domains-containing protein 3 and 1 (NLRP3, NLRP1) have been implicated in aging and neurodegenerative disease. Recent evidence suggests that NLRP3 and NLRP1 have gene regulatory effects that directly connect their assembly to the innate immune metabolism of microglia, presumably through the regulation of key elements of the Krebs cycle. In our project, we will investigate, using human iPSC-derived microglia as an experimental model, how NLRP1-mediated gene regulation influences (i) microglial transcriptomes and (ii) microglial function, which are key for maintaining synaptic integrity and neuronal plasticity. Since microglial senescence has been found to cause tau pathology, a major phenotype of the aging and degenerating brain, we will further study (iii) how NLRP1 affects microglial senescence and renewal in the respective models.
-
Project details (PDF):
-
Duration:
2023-2025
-
Funding source:
PI funding (University of Luxembourg)
-
Researchers:
Masanori ITAKURA
-
Partners:
-
Description:
Amyloid-beta (Aβ), α-synuclein, and TDP-43 are not only known to be etiologic molecules in Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), but have also been shown to undergo several post-translational modifications in response to their surrounding environment. Our project aims to investigate and compare the effects of post-translational modification (nitration, oxidation, glycation, etc.) of pathogenic molecules (Aβ, α-synuclein, TDP-43) on neuroinflammation and neurodegeneration. To achieve this, we will employ artificially modified or isolated molecules from brains of rodent disease models to test a) whether microglia can recognise and internalise these modified proteins, and b) whether and how these modified proteins affect the functionality of microglia (phenotypic change, cytokine production, clearance of aggregated proteins) in vitro and in vivo.
-
Project details (PDF):
-
Duration:
2022-2027
-
Funding source:
PI funding (University of Luxembourg)
-
Researchers:
Arnaud MARY
-
Partners:
-
Description:
Recent studies have uncovered neuroinflammation as a new crucial and early player in Alzheimer’s disease (AD), a discovery that has provided us with new potential treatment targets to study and test therapeutically. Pathological neuroinflammation in AD leads to pyroptosis, a pro-inflammatory mode of cell death that is mediated by inflammasomes and gasdermins. While the role of neuroinflammation in the development of AD is well documented, the importance of pyroptosis in this process still needs to be clarified. Hence, our project aims to better understand the mechanisms of microglial pyroptosis in the context of AD, and to test the relevance of inflammasome-dependent pyroptosis inhibition as a new strategy to counteract AD progression.
-
Project details (PDF):
-
Duration:
2024-2027
-
Funding source:
EU Joint Programme – Neurodegenerative Disease Research (JPND)
-
Researchers:
Wanda GRABON
-
Partners:
- Nicolas SERGEANT, Inserm LilNCog Research Center, University of Lille, France (Coordinator)
- Charlotte TEUNISSEN, University Medical Center Amsterdam, The Netherlands
- Ina VORBERG, German Center for Neurodegenerative Diseases (DZNE) Bonn, Germany
- Joelle VINGH, ESPCI CNRS, University Paris VI, France
- Krzysztof SOBCZAK, Adam Mickiewicz University, Poland
- Sermin GENC, Izmir Biomedicine and Genome Center, Turkey
-
Description:
Alzheimer’s disease (AD) is the most prevalent form of dementia, affecting tens of millions worldwide. Its pathophysiology is driven by the convergence of amyloid-beta (Aβ) deposition, neurofibrillary tangle formation from hyperphosphorylated tau, astrogliosis, and chronic neuroinflammation. Current drug development pipelines typically target only one of these processes, leaving a critical gap for true disease-modifying interventions. Through phenotypic screening, five families of small molecules have been developed that simultaneously mitigate amyloid pathology, tau aggregation, neuroinflammation, and cognitive decline in animal models (including the lead compound Ezeprogind, which has completed a phase IIa clinical trial). However, the precise biological targets and modes of action of these compounds remain undefined.
CCAD aims to identify these targets and decipher their mechanisms using an integrated multi-omics strategy. We will apply bioorthogonal chemistry (photoaffinity labelling, SuFEX, AUTOTAC clickable probes) coupled with chemoproteomics to capture and identify drug-target complexes. In parallel, we will map drug-induced molecular changes through deep RNA sequencing, spliceomics, single-cell and spatial transcriptomics (MERFISH), ATAC-sequencing, transcription factor footprinting, miRNA profiling, and proteomics, in cellular models of tau seeding and prion-like spreading and in Thy-Tau22 and APP/PS1 mouse models. The Luxembourg group will dissect drug effects on microglial inflammation, NLRP3 inflammasome activation, pyroptosis, and senescence-associated secretory phenotypes.
Big-data integration, performed at the BiLille platform, will combine these datasets with existing GWAS and biomarker cohorts to nominate druggable targets and clinically translatable diagnostic and theragnostic biomarkers, validated through Olink and Simoa immunoassays.
-
Project details (PDF):
-
Duration:
2024-2027
-
Funding source:
EU Joint Programme – Neurodegenerative Disease Research (JPND)
-
Researchers:
Itziar DE ROJAS
-
Partners:
Computational Biology research group (Prof. Antonio DEL SOL MESA)
-
Description:
Ageing is the strongest risk factor for Alzheimer’s disease (AD), and AD in turn dramatically accelerates cognitive deterioration. However, not every ageing individual develops dementia, suggesting that aging and AD operate through partially overlapping and partially independent biological pathways. Identifying processes that act at the intersection of these two trajectories would offer particularly attractive drug targets, as a single intervention could simultaneously address two major determinants of dementia. Existing research has often studied ageing and AD in isolation, leaving this convergence point poorly defined.
ADPriOMICs aims to disentangle shared and distinct molecular pathways using an integrative, multi-omics framework. We hypothesise that immune-related processes, particularly microglial activation, astrocyte–microglia crosstalk, and senescence-associated secretory phenotypes, represent a central biological bridge between aging and AD. To test this, we will combine CSF and plasma proteomics from 997 longitudinally-followed memory clinic patients, large-scale GWAS and transcriptome-wide association data from the EADB consortium, and whole-exome sequencing of nearly 50,000 participants enriched for extreme phenotypes (early-onset AD, late-onset AD survivors, and cognitively healthy individuals over 90). Bayesian informative hypothesis testing and protein-network analysis will rank candidate genes and proteins as ageing-specific, AD-specific, or shared.
Top candidates will be functionally validated in iPSC-derived 2D neurons, astrocytes, microglia, and 3D brain organoids using CRISPR/Cas9 engineering. Pharmacoepidemiological data from Danish population registries and UK Biobank will test whether marketed drugs targeting prioritised pathways modify dementia risk. Finally, predictive CSF and plasma biomarker signatures will be derived and validated in independent cohorts to support early dementia detection and patient stratification for future trials.
-
Project details (PDF):