
 Faculty of Science, Technology and Medicine
Faculty of Science, Technology and Medicine Faculty of Law, Economics and Finance
Faculty of Law, Economics and Finance Faculty of Humanities, Education and Social Sciences
Faculty of Humanities, Education and Social Sciences Interdisciplinary Centre for Security, Reliability and Trust
Interdisciplinary Centre for Security, Reliability and Trust Luxembourg Centre for Systems Biomedicine
Luxembourg Centre for Systems Biomedicine Luxembourg Centre for Contemporary and Digital History
Luxembourg Centre for Contemporary and Digital History Luxembourg Centre for European Law
Luxembourg Centre for European Law Luxembourg Centre for Socio-Environmental Systems
Luxembourg Centre for Socio-Environmental SystemsOur research
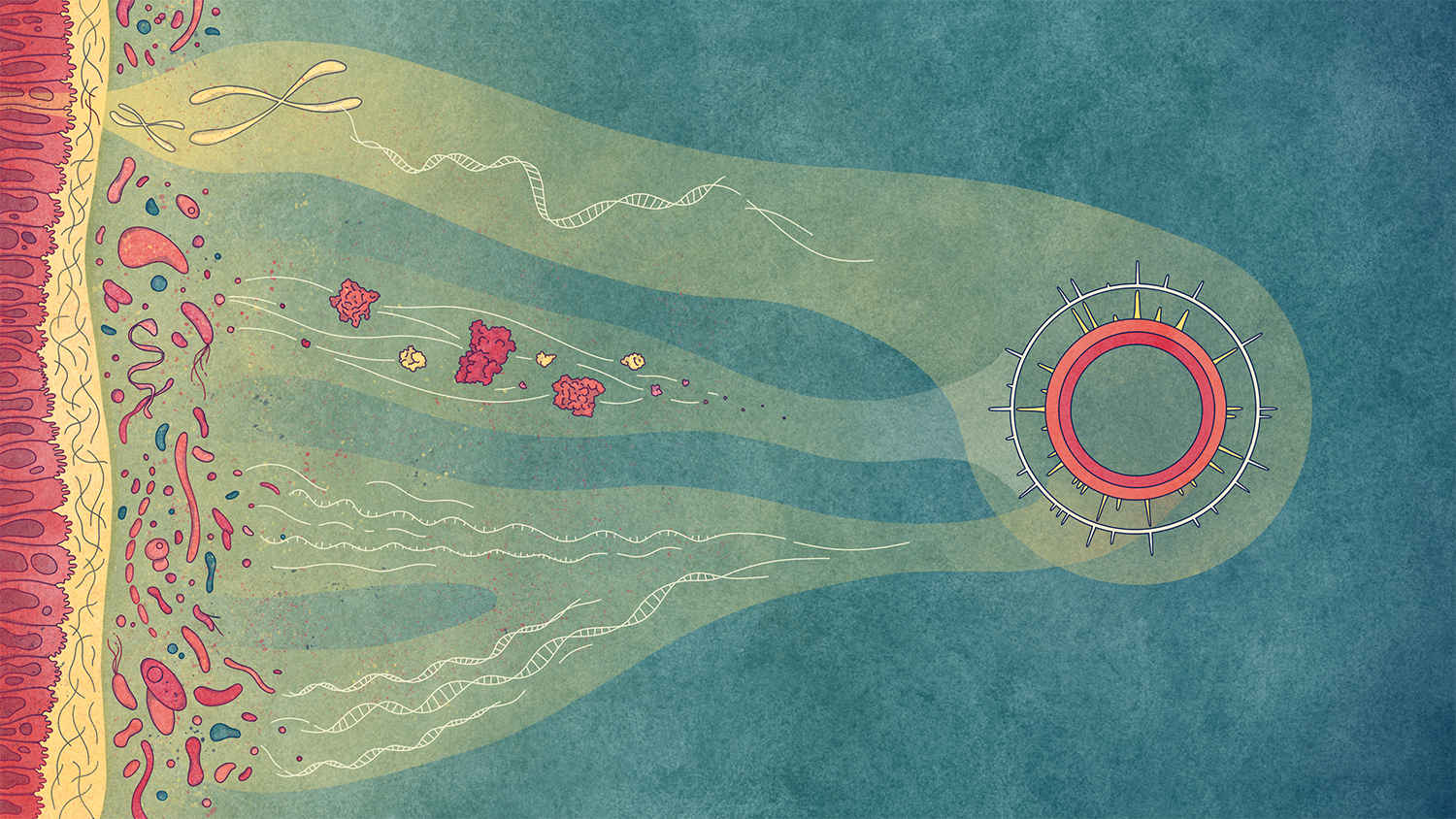
We are embedded in a microbial world. Naturally occurring microbial communities play fundamental roles in the Earth’s cycles and in human health. They provide essential services to mankind, removing contaminants from ecosystems for example. These highly dynamic and heterogeneous systems are at the heart of our research.
We use integrated omics to study complex microbial populations, especially human host-associated microbiota. The resulting data serves as input for the construction of multiscale models, allowing us to formulate and test new hypotheses. These can be validated using wet-lab experiments, including methods developed in our group. We have for example created a new microfluidics-based human-microbial co-culture device called HuMiX.
Ultimately, we hope to better understand mechanisms shaping complex microbiomes, with applications ranging from biodiesel to chronic diseases.
The Systems Ecology group was established under the auspices of the Luxembourg National Research Fund’s ATTRACT Programme.
Prof. Paul Wilmes has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant agreement No 863664).
Our research projects
The Systems Ecology group is involved in several research projects focusing on human diseases (colorectal cancer, diabetes, Parkinson’s), as well as on bacteria found in wastewater and biofilms:
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The human body is home to microbial communities whose member cells as well as genes outnumber our own. The largest part of the human microbiome resides in the gastrointestinal tract and it greatly influences human health and disease. Metagenomic DNA from faecal samples has been analysed in detail in recent years to understand the structure and function of the gastrointestinal microbiome. Links between colonization patterns and human well-being or disease have been identified. Here, we are using an integrated omics approach to identify potential links between gut microbial community structure and function, and colorectal cancers. In addition, differences/commonalities between the oral microbiome and the colonic microbiota are being analysed.
Collaborators: Peer Bork (European Molecular Biology Laboratory, Heidelberg, Germany), Serge Haan and Elisabeth Letellier (Life Sciences Research Unit, Luxembourg)
Funding: FNR CORE grant “microCancer”References:
- Ternes D, Tsenkova M, Pozdeev VI, Meyers M, Koncina E, Atatri S, Schmitz M, Karta J, Schmoetten M, Heinken A, Rodriguez F, Delbrouck C, Gaigneaux A, Ginolhac A, Nguyen TTD, Grandmougin L, Frachet-Bour A, Martin-Gallausiaux C, Pacheco M, Neuberger-Castillo L, Miranda P, Zuegel N, Ferrand JY, Gantenbein M, Sauter T, Slade DJ, Thiele I, Meiser J, Haan S, Wilmes P, Letellier E. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nature Metabolism, 2022, 4(4):458-475. doi: 10.1038/s42255-022-00558-0.
- Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in Colorectal Cancer: How to Get from Meta-omics to Mechanism? Trends in Microbiology, 2020, 28(5):401-423. doi: 10.1016/j.tim.2020.01.001.
- Schmidt TS, Hayward MR, Coelho LP, Li SS, Costea PI, Voigt AY, Wirbel J, Maistrenko OM, Alves RJ, Bergsten E, de Beaufort C, Sobhani I, Heintz-Buschart A, Sunagawa S, Zeller G, Wilmes P, Bork P. Extensive transmission of microbes along the gastrointestinal tract. Elife, 2019, 8:e42693, doi: 10.7554/eLife.42693
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Recent evidence from high-resolution molecular studies suggests links between microbial dysbiosis in the gastrointestinal tract and several complex chronic diseases including diabetes mellitus. Within our Luxembourg-based Diabetes Multiplex Family Studies (MUST), we investigate the interplay of the gastrointestinal microbiota, lifestyle and genetic background in a study of eight families with two or more individuals affected by type 1 diabetes mellitus. Faecal and saliva samples which have undergone comprehensive biomolecular extractions are analysed using a multi-omics approach yielding high-resolution, high-throughput molecular data. Anthropometric data, including demographics, medical history, health status, medication, and dietary habits of all study participants were collected and are integrated with molecular data.
Collaborators: Carine de Beaufort (LCSB), Karsten Hiller (TU Braunschweig), Fay Betsou (Integrated BioBank of Luxembourg), Dörte Becher (University Greifswald), and Peer Bork (EMBL Heidelberg)
References:
- Kunath BJ, Hickl O, Queirós P, Martin-Gallausiaux C, Lebrun LA, Halder R, Laczny CC, Schmidt TSB, Hayward MR, Becher D, Heintz-Buschart A, de Beaufort C, Bork O, May P, Wilmes P. Alterations of oral microbiota and impact on the gut microbiome in type 1 diabetes mellitus revealed by multi-omic analysis. Microbiome, 2022. doi: 10.1101/2022.02.13.480246
- Heintz-Buschart A, May P, Laczny CC, Lebrun LA, Bellora C, Krishna A, Wampach L, Schneider JG, Hogan A, de Beaufort C, Wilmes P. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nature Microbiology, 2016, 2:16180. doi: 10.1038/nmicrobiol.2016.180
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Perturbations to the colonisation process of the human gastrointestinal tract induced by caesarean section delivery have been suggested to result in adverse health effects later in life. Although much research has been performed on bacterial colonization and succession, much less is known about the other two domains of life, archaea and eukaryotes and their potential impact on neonatal health. Based on previous results, we observed that fundamental differences in the microbiome were observable as early as 3 days after birth between delivery modes (vaginal vs caesarean section delivery). However, more specific information on the earliest time after delivery and the potential transfer of neonatal colonizers from mother to infant is proportionally low to date.
In order to detect and analyse the colonization process over the first year of life in vaginally and caesarean section delivered infants, a longitudinally sampled study called Cosmic has been set up in Luxembourg. The study is divided into 2 subsections, one focussing on the colonization of the neonatal gastrointestinal microbiome by all 3 domains of life (bacteria, archaea and eukaryotes), while the follow-up study mainly targets the earliest colonisation process and the potential transfer of neonatal colonisers from mother to infant using high-throughput techniques and specifically adapted methods to minimize the impact of potential contaminants.
Collaborators: Carine de Beaufort (CHL) ; Integrated BioBank of Luxembourg (IBBL)
Funding: FNR AFR – Colonisation, succession and evolution of the human gastrointestinal microbiome in infants at high risk of metabolic disease in adulthood – COSMIC-PhD; Fondation André et Henriette Losch; FNR PRIDE – Microbiomes in One HealthReferences:
- Busi SB, de Nies L, Habier J, Wampach L, Fritz JV, Heintz-Buschart A, May P, Halder R, de Beaufort C, Wilmes P. Persistence of birth mode-dependent effects on gut microbiome composition, immune system stimulation and antimicrobial resistance during the first year of life. ISME Communications, 2021, 1:8, doi: 10.1038/s43705-021-00003-5.
- Wampach L, Heintz-Buschart A, Fritz JV, Ramiro-Garcia J, Habier J, Herold M, Narayanasamy S, Kaysen A, Hogan AH, Bindl L, Bottu J, Halder R, Sjöqvist C, May P, Andersson AF, de Beaufort C, Wilmes P. Birth mode is associated with earliest strain-conferred gut microbiome functions and immunostimulatory potential. Nature Communications, 2018, 9(1):5091, doi: 10.1038/s41467-018-07631-x.
- Wampach L, Heintz-Buschart A, Hogan A, Muller EEL, Narayanasamy S, Laczny CC, Hugerth LW, Bindl L, Bottu J, Andersson AF, de Beaufort C, Wilmes P. Colonization and Succession within the Human Gut Microbiome by Archaea, Bacteria, and Microeukaryotes during the First Year of Life. Frontiers in Microbiology, 2017, 8:738, doi: 10.3389/fmicb.2017.00738
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Parkinson’s disease (PD) is a neurodegenerative disease with characteristic motor symptoms that are commonly accompanied by pathological α-synuclein aggregation. Presently the propagation of α-synuclein aggregation in PD is proposed to start in the periphery, i.e. in the enteric nervous system and the olfactory bulb. We hypothesize that changes in microbial community structure and function accompany PD from its onset and progression through its most specific prodrome, idiopathic REM sleep behaviour disorder (iRBD), to manifest PD. In MiBiPa, we use an integrated multi-omic approach to study the connections of PD and microbiota based on several types of samples (faeces, nasal washes and early morning saliva) from patients with PD and iRBD as well as healthy control subjects. The overall aims of the project include discovery of early-stage biomarkers for PD, and contributing to the overall understanding of how microbiota influence the development of the disease.
Our gut microbiota results have so far revealed that differences like those seen between PD patients and control subjects can also be detected in the prodromal iRBD stage. Furthermore, we have discovered a compound produced by gut archaea that could play an important role in the pathogenesis of PD. In addition to the case-control studies, the MiBiPa-related subprojects include a dietary intervention study (PARKdiet) where we explore whether a fibre-supplemented diet could alleviate the symptoms of PD via beneficial alterations of the gut microbiome. Our extensive work in the context of PD and microbiota is of particular relevance in relation to the development of future disease-modifying neuroprotective therapies that would require intervention at the earliest stages of disease but also to identify preventive strategies for PD.
Collaborators: Brit Mollenhauer (Paracelsus Elena Klinik Kassel, Germany), Wolfgang Oertel (Philipps-University Marburg)Funding: FNR CORE (MiBiPa), Michael J. Fox Foundation (MiBiPa-PLUS and PARKdiet), Parkinson’s Foundation (MiBiPa Saliva)
References:
- Heintz-Buschart A, Pandey U, Wicke T, Sixel-Döring F, Janzen A, Sittig-Wiegand E, Trenkwalder C, Oertel WH, Mollenhauer B, Wilmes P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord, 2018. 33(1):88–98. doi: 10.1002/mds.27105
- Trezzi J-P, Aho V, Jäger C, Schade S, Janzen A, Hickl O, Kunath B, Thomas M, Schmit K, Garcia P, Sciortino A, Martin-Gallausiaux C, Halder R, Uriarte Huarte O, Heurtaux T, Heins-Marroquin U, Gomez-Giro G, Weidenbach K, Delacour L, Laczny C, Novikova P, Ramiro-Garcia J, Singh R, Talavera Andújar B, Lebrun L, Daujeumont A, Habier J, Dong X, Gavotto F, Heintz-Buschart A, Schneider J, Jehmlich N, von Bergen M, Schymanski E, Schmitz R, Schwamborn J, Glaab E, Linster C, Kitami T, Buttini M, May P, Trenkwalder C, Oertel W, Mollenhauer B, Wilmes, P. An archaeal compound as a driver of Parkinson’s disease pathogenesis. ResearchSquare preprint, 2022, doi: 10.21203/rs.3.rs-1827631/v1
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The human gastrointestinal tract is densely colonised by several bacterial species, and imbalances in the microbiota influence human health. Bacteria are known to change expression of genes to cope with modified environments and bacterial sRNAs play crucial roles in such responses. Furthermore, several studies have postulated that bacterial sRNAs influence bacterial pathogenesis. In the present projects, we are characterising the functions of sRNAs (primarily exported sRNAs) which we have identified in the gastrointestinal tract and in enteric bacteria to understand whether these molecules may play roles in interspecies communication.
Collaborators: Esther N.M. Nolte-‘t Hoen (Utrecht University, Dept Biochemistry & Cell Biology, Fac Veterinary Medicine, Netherlands); Julien Godet (Laboratoire de Biophotonique et Pharmacologie, UMR CNRS 7213, Faculté de Pharmacie)
Funding: BEaR FNR COREJunior programme grantReferences:
- Martin-Gallausiaux C, Malabirade A, Habier J, Wilmes P. Fusobacterium nucleatum Extracellular Vesicles Modulate Gut Epithelial Cell Innate Immunity via FomA and TLR2. Frontiers in Immunology, 2020,11:583644. doi: 10.3389/fimmu.2020.583644.
- Malabirade A, Habier J, Heintz-Buschart A, May P, Godet J, Halder R, Etheridge A, Galas D, Wilmes P, Fritz JV. The RNA Complement of Outer Membrane Vesicles From Salmonella enterica Serovar Typhimurium Under Distinct Culture Conditions. Frontiers in Microbiology, 2018, 9:2015. doi: 10.3389/fmicb.2018.02015.
- Ghosal A, Upadhyaya BB, Fritz JV, Heintz-Buschart A, Desai MS, Yusuf D, Huang D, Baumuratov A, Wang K, Galas D, Wilmes P. The extracellular RNA complement of Escherichia coli. Microbiologyopen. 2015 Jan 21. doi: 10.1002/mbo3.235.
- Joëlle V. Fritz; Extracellular RNA in Bacteria
http://exrna.org/extracellular-rna-bacteria/
- Fritz, J. V., Heintz-Buschart, A., Ghosal, A., Wampach, L., Etheridge, A., Galas, D., & Wilmes, P. (2016). Sources and Functions of Extracellular Small RNAs in Human Circulation. Annual review of nutrition. doi:10.1146/annurev-nutr-071715-050711
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The diverse ecology of the human gut microbiome is vital to human physiology. Numerous inflammatory-based chronic disorders, such as autoimmune and neurological diseases, are linked to changes in the microbiome. Chronic disorders, by definition, develop over longer time and require continued medical attention. This directly or indirectly reduces the quality of life of the affected individuals and possibly of their families and friends. The presence or imbalance of specific microorganisms (“who is there”) are not the only disease-related changes, however. High amounts of effector molecules produced by the microbiome, such as nucleic acids, (poly)peptides, and metabolites, are found in the gut. These molecules have thus far not been thoroughly studied. This information gap restricts the mechanistic understanding of the microbiome’s functional impact on chronic disorders including Parkinson’s disease and rheumatoid arthritis. The ExpoBiome project aims to, for the first time, comprehensively identify the components of this effector molecule complex and their effects on the human immune system. The project will develop and apply cutting-edge molecular approaches on microbiome samples taken from healthy people and those who have just been diagnosed with PD or RA. The resulting data will be integrated using existing and newly developed computational biology and machine learning approaches to put it into existing context and generate new insights on microbial factors in health and disease. A model clinical intervention (fasting) with the aim to reduce inflammation and thereby improving health will be applied and the resulting changes of the microbiome will be reviewed. Newly identified anti-inflammatory compounds will be specifically studied using a gut-on-chip model. ExpoBiome will thus lead to key advances to better understand how microbiome shifts provide benefits in the context of inflammatory-based chronic disordered, how specific molecules can be used to improve health, and to forecast treatment outcomes, thereby substantially contributing to the development of future diagnostic and therapeutic applications.
Collaborators: Brit Mollenhauer (Paracelsus Elena Klinik Kassel, Germany), Andreas Michalsen and Thomas Häupl (Charité Clinical Center Berlin, Germant), Robert Hettich (Oak Ridge National Laboratory, USA)
Funding: European Research Council Consolidator Grant “Deciphering the impact of exposures from the gut microbiome-derived molecular complex in human health and disease”.
References:
- De Saedeleer B, Malabirade A, Ramiro-Garcia J, Habier J, Trezzi J-P, Peters SL, Daujeumont A, Halder R, Jäger C, Busi SB, May P, Oertel W, Mollenhauer B, Laczny CC, Hettich RL, Wilmes P. Systematic characterization of human gut microbiome-secreted molecules by integrated multi-omics. ISME Communications 2021, 1, 1–6, doi: 10.1038/s43705-021-00078-0.
- Wilmes P, Martin-Gallausiaux C, Ostaszewski M, Aho VTE, Novikova PV, Laczny CC, Schneider JG. The gut microbiome molecular complex in human health and disease. Cell Host & Microbe 2022, 30, 1201–1206, doi: 10.1016/j.chom.2022.08.016.
- Aho VTE, Ostaszewski M, Martin-Gallausiaux C, Laczny CC, Schneider JG, Wilmes P. SnapShot: The Expobiome Map. Cell Host & Microbe, 2022, 30, 1340-1340.e1, doi: 10.1016/j.chom.2022.08.015.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
This project involves the reconstruction of bacterial genomes isolated from our model wastewater lipid accumulating communities. The lack of genome sequences from appropriate reference organisms continues to hamper the integration of community-level multi-omic data (e.g., metagenomic, metatranscriptomic, metaproteomic, etc) from all but the most heavily studied microbial ecosystems. To address this limitation, we have sequenced the genome of Candidatus Microthrix parvicella, a model lipid-accumulating bacterium; and are working on sequencing that of 130 additional isolates. The genome sequences will help us improve our assembly and read-recruitment analyses of community sequence data. Coupled with phenotypic characterization of these organisms, they will also give us an insight into the biology of the dominant members of our model community.
Collaborators: Paul Keim and Lance Price (TGen North, Flagstaff, Arizona, USA)
Funding: FNR ATTRACT programme “SysBioNaMA”References:
- Muller EEL, Narayanasamy S, Zeimes M, Laczny CC, Lebrun LA, Herold M, Hicks ND, Gillece JD, Schupp JM, Keim P, Wilmes P. First draft genome sequence of a strain belonging to the Zoogloea genus and its gene expression in situ. Standards in Genomic Sciences, 2017, 12:64. doi: 10.1186/s40793-017-0274-y.
- Muller EEL, Pinel N, Gillece JD, Schupp JM, Price LB, Engelthaler DM, Levantesi C, Tandoi V, Luong K, Baliga NS, Korlach J, Keim PS, Wilmes P. Genome Sequence of “Candidatus Microthrix parvicella” Bio17-1, a Long-Chain-Fatty-Acid-Accumulating Filamentous Actinobacterium from a Biological Wastewater Treatment Plant. Journal of Bacteriology, 2012, 194:6670–6671, doi: 10.1128/JB.01765-12.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
In this project, we are characterizing microbial communities at the genomic and transcriptomic level, with a special interest on lipid accumulating bacterial populations, which are naturally enriched in biological wastewater treatment systems and may be harnessed for the conversion of mixed lipid substrates (wastewater) into biodiesel. We explicitly aim to elucidate the genetic blueprints and the functional relevance of specific populations within the community. We are focusing on within-population genetic and functional heterogeneity, trying to understand how fine-scale variations contribute to differing lipid accumulating phenotypes. Insights from this project will help us understand at a fundamental level the functioning of microbial ecosystems; and in a concrete level, help us improve optimisation and modeling strategies for current and future biological wastewater treatment processes.
Collaborators: Paul Keim and Lance Price (TGen North, Flagstaff, Arizona, USA)
Funding: FNR ATTRACT programme “SysBioNaMA”; MetaLABpop FNR AFR PDR programme grant.References:
- Muller EE, Glaab E, May P, Vlassis N, Wilmes P. Trends Microbiol. 21:325-333, doi: 10.1016/j.tim.2013.04.009.
- Muller EE, Pinel N, Laczny C, Hoopmann M, Narayanasamy S, Lebrun LA; Roume H, Lin J, May P, Hicks N, Buschart A, Wampach L, Liu C, Price L, Gillece J, Guignard C, Schupp J, Vlassis N, Baliga N, Moritz R, Keim P, Wilmes P. Nature Communications, 2014, 5:5603, doi: 10.1038/ncomms6603
- Roume H, Heintz-Buschart A, Muller EEL, May P, Satagopam VP, Laczny CC, Narayanasamy S, Lebrun LA, Hoopmann MR, Schupp JM, Gillece JD, Hicks ND, Engelthaler DM, Sauter T, Keim PS, Moritz RL, Wilmes P. Comparative integrated omics: identification of key functionalities in microbial community-wide metabolic networks. npj Biofilms and Microbiomes, 2015, 1, 15007, doi: 10.1038/npjbiofilms.2015.7
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Environmentally friendly techniques, such as biomining, must be developed to meet the increased European demand for metals. Biomining exploits acidophilic microorganisms for the recovery of metals from sulphide ores in tanks, heaps and dumps. In this project, biofilm formation of moderately thermophilic bioleaching bacteria, Acidithiobacillus caldus, Leptospirillum ferriphilum and Sulfobacillus thermosulfidooxidans on chalcopyrite surfaces will be studied with the aim to improve leaching of copper from the mineral. Since a unique feature of the experimental setup is the use of well-defined microbial communities of limited diversity and known cultivation conditions, it will be possible to establish and test novel approaches for measuring and modelling a mixed-microorganism biofilm formation process.
Collaborators: Mark Dopson, Stephan Christel (Linneaus University Kalmar), Ansgar Poetsch (Ruhr Universitaet Bochum), Wolfgang Sand, Mario Vera (Universitaet Duisburg Essen), Igor Pivkin, Antoine Buetti-Dinh (Università della Svizzera italiana)
Funding: EU ERASysApp grant “SysMetEx”References:
- Christel S, Herold M, Bellenberg S, El Hajjami M, Buetti-Dinh A, Pivkin IV, Sand W, Wilmes P, Poetsch A, Dopson M. Multi-omics Reveals the Lifestyle of the Acidophilic, Mineral-Oxidizing Model Species Leptospirillum ferriphilumT. Applied and Environmental Microbiology, 2018, 84(3):e02091-17. doi: 10.1128/AEM.02091-17.
- Buetti-Dinh A, Galli V, Bellenberg S, Ilie O, Herold M, Christel S, Boretska M, Pivkin IV, Wilmes P, Sand W, Vera M, Dopson M. Deep neural networks outperform human expert’s capacity in characterizing bioleaching bacterial biofilm composition. Biotechnology Reports, 2019, 22:e00321. doi: 10.1016/j.btre.2019.e00321.
- Bellenberg S, Buetti-Dinh A, Galli V, Ilie O, Herold M, Christel S, Boretska M, Pivkin IV, Wilmes P, Sand W, Vera M, Dopson M. Automated Microscopic Analysis of Metal Sulfide Colonization by Acidophilic Microorganisms. Applied and Environmental Microbiology, 2018, 84(20):e01835-18. doi: 10.1128/AEM.01835-18.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The project aims to elucidate details of the fate of lipids within mixed microbial communities present at the surface of an anoxic activated sludge wastewater treatment system, in view of potentially harnessing these traits for direct production of biodiesel from wastewater. However, such mixed microbial communities exhibit extensive complex and various interactions and we still know very little about their overall functional capacity. In this project, we use a unique framework for comparative eco-systems biology in which we integrate multi-omics data including metagenomic, metatranscriptomic, metaproteomic and metabolomic information into a community-level metabolic network reconstruction. The mapping of the multi-omic information on the network allows us to identify key genes involved in conferring the community-wide lipid accumulation phenotype. The developed approach is further applicable to other microbial communities for which dynamic omic data exists
Funding: FNR ATTRACT programme grant ”SysMetEx”; SysBioWwTBioEng FNR AFR PHD programme grantReferences:
- Herold M, Martínez Arbas S, Narayanasamy S, Sheik AR, Kleine-Borgmann LAK, Lebrun LA, Kunath BJ, Roume H, Bessarab I, Williams RBH, Gillece JD, Schupp JM, Keim PS, Jäger C, Hoopmann MR, Moritz RL, Ye Y, Li S, Tang H, Heintz-Buschart A, May P, Muller EEL, Laczny CC, Wilmes P. Integration of time-series meta-omics data reveals how microbial ecosystems respond to disturbance. Nature Communications, 2020, 1(1):5281. doi: 10.1038/s41467-020-19006-2.
- Martínez Arbas S, Narayanasamy S, Herold M, Lebrun LA, Hoopmann MR, Li S, Lam TJ, Kunath BJ, Hicks ND, Liu CM, Price LB, Laczny CC, Gillece JD, Schupp JM, Keim PS, Moritz RL, Faust K, Tang H, Ye Y, Skupin A, May P, Muller EEL, Wilmes P. Roles of bacteriophages, plasmids and CRISPR immunity in microbial community dynamics revealed using time-series integrated meta-omics. Nature Microbiology, 2021, 6(1):123-135. doi: 10.1038/s41564-020-00794-8.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Advances towards efficiency improvement of the anaerobic digestion process (AD) for biogas production is often quoted to be dependent on two major key subjects of research (1) an in depth understanding of the structure and dynamics of the microbial populations involved in the process and (2) the development of on-line monitoring tools to better predict process dysfunction occurring during inadequate organic loading rate. We are applying our integrated omics methodology to obtain detailed insights into the microbial ecology and microbial community dynamics of the anaerobic digestion process in relation to reactor design and feeding regime. We hope to identify biomarkers which will prove helpful for real-time performance monitoring of AD plants.
Collaborator: Philippe Delfosse (Centre de Recherche Public – Gabriel Lippmann, Belvaux, Luxembourg)
Funding: FNR CORE programme grant “GASPOP” CLOMICS
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Several associations have been established between the observed microbial community structures within activated sludge and specific factors. Factors which have been identified as drivers of community structure include: (i) immigration from influent wastewater, (ii) species-species interactions, (iii) wastewater composition, (iv) operational parameters and/or environmental conditions, (v) severe perturbations to plant operation, and (vi) seasonality. At present, it is difficult to conclusively determine which, or which combinations thereof, are the most important factors governing microbial community structure not least because there is conflicting evidence in the literature. We have continuously sampled the OMMCs on a weekly basis and have now collected a time series which covers five years including the corresponding abiotic factors. We will leverage this unprecedented longitudinal dataset by generating and analysing high-resolution and high-fidelity multi-omic data together with operational and environmental data to determine the main biotic factors, operational parameters and environmental conditions which govern the dynamics of OMMCs in a full-scale biological WWTP.
Collaborators: Emilie Muller (Université de Strasbourg, France), Stephanie Widder (Medical University of Vienna, Austria).
Funding: FNR CORE “GoMiCo”
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The melting of the cryosphere is among the most conspicuous consequences of climate change, with impacts on microbial life and related biogeochemistry. However, we are missing a systematic understanding of microbiome structure and function across cryospheric ecosystems. Here, we have established a global inventory of the microbiome from snow, ice, permafrost soils, and both coastal and freshwater ecosystems under glacier influence. Combining phylogenetic and taxonomic approaches, we find that these cryospheric ecosystems, despite their particularities, share a microbiome with representatives across the bacterial tree of life and apparent signatures of early and constrained radiation. Our findings reveal key genomic underpinnings of adaptive traits contributing to the success of complex biofilms to exploit environmental opportunities especially in glacier-fed streams, which are now rapidly changing owing to global warming. Importantly, this work provides a reference resource for future studies on climate change microbiology.
Collaborator: Tom Battin (EPFL, Switzerland)
Funding: Swiss National Science Foundation “ENSEMBLE” Synergia grant.References:
- Brandani J, Peter H, Busi SB, Kohler TJ, Fodelianakis S, Ezzat L, Michoud G, Bourquin M, Pramateftaki P, Roncoroni M, Lane SN, Battin TJ. Spatial patterns of benthic biofilm diversity among streams draining proglacial floodplains. Frontiers in Microbiology, 2022, 13:948165, doi: 10.3389/fmicb.2022.948165.
- Ezzat L, Fodelianakis S, Kohler TJ, Bourquin M, Brandani J, Busi SB, Daffonchio D, De Staercke V, Marasco R, Michoud G, Oppliger E, Peter H, Pramateftaki P, Schön M, Styllas M, Tadei V, Tolosano M, Battin TJ. Benthic Biofilms in Glacier-Fed Streams from Scandinavia to the Himalayas Host Distinct Bacterial Communities Compared with the Streamwater. Applied Environmental Microbiology, 2022, 88(12):e0042122, doi: 10.1128/aem.00421-22.
- Bourquin M, Busi SB, Fodelianakis S, Peter H, Washburne A, Kohler TJ, Ezzat L, Michoud G, Wilmes P, Battin TJ. The microbiome of cryospheric ecosystems. Nature Communications, 2022, 13(1):3087, doi: 10.1038/s41467-022-30816-4.
- Kohler TJ, Fodelianakis S, Michoud G, Ezzat L, Bourquin M, Peter H, Busi SB, Pramateftaki P, Deluigi N, Styllas M, Tolosano M, de Staercke V, Schön M, Brandani J, Marasco R, Daffonchio D, Wilmes P, Battin TJ. Glacier shrinkage will accelerate downstream decomposition of organic matter and alters microbiome structure and function. Global Change Biology, 2022, 28(12):3846-3859, doi: 10.1111/gcb.16169.
- Busi SB, Bourquin M, Fodelianakis S, Michoud G, Kohler TJ, Peter H, Pramateftaki P, Styllas M, Tolosano M, De Staercke V, Schön M, de Nies L, Marasco R, Daffonchio D, Ezzat L, Wilmes P, Battin TJ. Genomic and metabolic adaptations of biofilms to ecological windows of opportunity in glacier-fed streams. Nature Communications, 2022, 13(1):2168, doi: 10.1038/s41467-022-29914-0.
- Fodelianakis S, Washburne AD, Bourquin M, Pramateftaki P, Kohler TJ, Styllas M, Tolosano M, De Staercke V, Schön M, Busi SB, Brandani J, Wilmes P, Peter H, Battin TJ. Microdiversity characterizes prevalent phylogenetic clades in the glacier-fed stream microbiome. The ISME Journal, 2022, 16(3):666-675, doi: 10.1038/s41396-021-01106-6.
- Kohler TJ, Peter H, Fodelianakis S, Pramateftaki P, Styllas M, Tolosano M, de Staercke V, Schön M, Busi SB, Wilmes P, Washburne A, Battin TJ. Patterns and Drivers of Extracellular Enzyme Activity in New Zealand Glacier-Fed Streams. Frontiers in Microbiology, 2020, 11:591465, doi: 10.3389/fmicb.2020.591465.
- Busi SB, Pramateftaki P, Brandani J, Fodelianakis S, Peter H, Halder R, Wilmes P, Battin TJ. Optimised biomolecular extraction for metagenomic analysis of microbial biofilms from high-mountain streams. PeerJ, 2020, 8:e9973. doi: 10.7717/peerj.9973.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
We are building a culture collection of lipid accumulating bacteria isolated from biological wastewater treatment plants under various different culture conditions. Isolates are screened for the lipid accumulation phenotype before undergoing whole genome sequencing. 600 isolates have been obtained so far which are currently undergoing in-depth characterisation.
Funding: FNR ATTRACT programme grant “SysBioNaMA”; MetaLABpop FNR AFR PDR programme grant.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
In microbial ecology, high-resolution molecular biology approaches are vital for discovering and characterising the vast microbial diversity, and understanding the interaction of microbial communities with biotic and abiotic environmental factors. Integrated omics, comprising community genomics, transcriptomics, proteomics and metabolomics, is able to reveal the links between genetic potential and functionality in microbial communities in a truly systematic fashion. However, mixed microbial communities are complex, dynamic and heterogeneous and it is therefore essential that biomolecular fractions obtained for high-throughput omic analyses are representative of single undivided samples to facilitate meaningful data integration, analysis and modelling. We have developed a new methodological framework for the reproducible sequential isolation of high-quality polar and non-polar metabolites polar and non-polar, RNA (optionally split into large and small RNA fractions), DNA and proteins from single undivided mixed microbial community samples. The developed methodological framework lays the foundation for standardised molecular eco-systematic studies on a range of different microbial communities in the future.
Funding: FNR ATTRACT programme grant “SysBioNaMA”; SysBioWwTBioEng FNR AFR PHD programme grant; MetaLABpop FNR AFR PDR programme grant; European Union Joint Programme – Neurodegenerative Disease Research grant.References:
- De Saedeleer B, Malabirade A, Ramiro-Garcia J, Habier J, Trezzi J-P, Peters SL, Daujeumont A, Halder R, Jäger C, Busi SB, May P, Oertel W, Mollenhauer B, Laczny CC, Hettich RL, Wilmes P. Systematic characterization of human gut microbiome-secreted molecules by integrated multi-omics. ISME Communications 2021, 1, 1–6, doi: 10.1038/s43705-021-00078-0.
- Roume H, EL Muller E, Cordes T, Renaut J, Hiller K & Wilmes P. A biomolecular isolation framework for eco-systems biology. The ISME Journal, 2013, 7: 110–121. doi: 10.1038/ismej.2012.72.
- Roume H, Heintz-Buschart A, Muller EE, & Wilmes P. Sequential isolation of metabolites, RNA, DNA, and proteins from the same unique sample. Methods Enzymology, 2013, 531: 219-236, doi: 10.1016/B978-0-12-407863-5.00011-3.
- Muller EE, Heintz-Buschart A, Roume H, Wilmes P. The sequential isolation of metabolites, RNA, DNA, and proteins from a single, undivided mixed microbial community sample. Protocol Exchange, 2013, doi: 10.1038/protex.2014.051.
- Pranjul S, Muller EE, Lebrun LA, Wampach L, Wilmes P. Sequential isolation of DNA, RNA, protein and metabolite fractions from murine organs and intestinal contents for integrated omics of host-microbiota interactions. Methods in Molecular Biology.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Integrated omic analysis of biological samples aims to resolve the information within each biomolecular fraction from genetic potential of the organisms to their functional capacity. Our comprehensive biomolecular isolation protocol lays the foundation for systematic integrated omics. The protocol itself is time consuming and therefore throughput is mediocre. Consequently, we are investigating approaches for automation of the biomolecular extraction protocol to improve throughput and provide a platform for standardized integrated omic analyses of different biological samples in the future.
Funding: FNR ATTRACT programme grant “SysBioNaMA”; MetaLABpop FNR AFR PDR programme grant; European Union Joint Programme – Neurodegenerative Disease Research grant.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Comprehensive analyses of biomolecules from low input samples, for example sRNAseq from human plasma samples, are prone to distortion by different biases. The danger of contamination with biomaterial from lab reagents is imminent. In this project, we develop strategies to control the cleanness of lab reagents and to improve the safety of commonly used extraction kits.
Collaborators: David Galas (Pacific Northwest Diabetes Research Institute, Seattle, USA), QiagenReference:
- Heintz-Buschart A, Yusuf D, Kaysen A, Etheridge A, Fritz JV, May P, de Beaufort C, Upadhyaya BB, Ghosal A, Galas DJ, Wilmes P. Small RNA profiling of low biomass samples: identification and removal of contaminants. BMC Biology, 2018, 16(1):52. doi: 10.1186/s12915-018-0522-7.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Any environmental sample, including those taken from human subjects (e.g. from the gastrointestinal tract) contain a complex mixture of microbial organisms. Due to current limitations of isolated culturing of many of these organisms, community genomics are generally used to study microbial consortia in situ. However, the scrambled genomic information obtainable using metagenomics needs to be related back to the organisms of origin, i.e. binned, so as to be able to study the individual community members separately. The advent of next-generation sequencing allows to create very large data sets, hence further increasing the difficulty of processing these complex mixtures. The goal of this computational biology project is to use in silico approaches for the retrieval of the individual sequences of the constituent organisms in an efficient, precise and automated way using state-of-the-art machine learning approaches. Downstream applications (e.g. genomic assembly and multi-omics integration) will benefit from this step by, among others, reducing the complexity, increasing sensitivity and allowing for parallel workflows.
Funding: DissRNA FNR AFR PHD programme grant (to Cédric Laczny), FNR PRIDE – Microbiomes in One HealthReferences:
- Hickl O, Queirós P, Wilmes P, May P, Heintz-Buschart A. binny: an automated binning algorithm to recover high-quality genomes from complex metagenomic datasets, bioRxiv preprint, 2021, doi: 10.1101/2021.12.22.473795.
- Laczny, C. C., Pinel, N., Vlassis, N., & Wilmes, P. (2014). Alignment-free visualisation of metagenomic data by nonlinear dimension reduction. Scientific reports, 4, 4516. doi:10.1038/srep04516
- Laczny, C. C., Sternal, T., Plugaru, V., Gawron, P., Atashpendar, A., Margossian, H. H., et al. (2015). VizBin – an application for reference-independent visualisation and human-augmented binning of metagenomic data. Microbiome, 3(1), 1. doi:10.1186/s40168-014-0066-1
- Laczny, C. C., Muller, E. E. L., Heintz-Buschart, A., Herold, M., Lebrun, L. A., Hogan, A., et al. (2016). Identification, Recovery, and Refinement of Hitherto Undescribed Population-Level Genomes from the Human Gastrointestinal Tract. Frontiers in microbiology, 7(75), 533. doi:10.1089/10665270050081478
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Molecular Eco-Systems Biology generates massive omics datasets derived from next generation sequencing and high-throughput mass spectrometry. This data has to be filtered, processed and analysed in an integrated fashion. We are developing methods which allow integration of metagenomic, metatranscriptomic, metaproteomic and metabolomic data by combining efficient in-house pipelines together with state-of-the-art publicly available software tools.
Funding: FNR ATTRACT programme grant “SysBioNaMA”; FNR AFR PHD programme grant to Shaman Narayanasamy.Reference:
- Narayanasamy S, Jarosz Y, Muller EE, Heintz-Buschart A, Herold M, Kaysen A, Laczny CC, Pinel N, May P, Wilmes P. IMP: a pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biology, 2016, 17(1):260, doi: 10.1186/s13059-016-1116-8
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Resolving microbial communities to study their microbial ecology requires to reconstruct the genomes of the individual community members to infer the taxonomic composition and the functional potential. The emergence of long read sequencing provides distinct advantages for genomic reconstruction, a.k.a., genome assembly, and thus helps to overcome limitations of short read sequencing, e.g., highly fragmented assemblies. However, approaches to evaluate the assembly quality on real-world data, i.e., without reference genomes, are lacking. Using functional meta-omic data, notably metatranscriptomics and metaproteomics, we show that different assembly algorithms reconstruct unique genes. Some of these genes are transcribed (metatranscriptomics) or translated (metaproteomics), thus showing that individual assembly algorithms produce partial results and that functional meta-omic data can be to identify high-confidence gene reconstructions, thereby serving as a proxy for an internal standard.
Reference:
- Galata V, Busi SB, Kunath BJ, de Nies L, Calusinska M, Halder R, May P, Wilmes P, Laczny CC. Functional meta-omics provide critical insights into long- and short-read assemblies. Briefings in Bioinformatics, 2021, 22(6):bbab330. doi: 10.1093/bib/bbab330.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Metaproteomics has emerged as the ideal approach to understand the effect of the microbiome-derived protein complement. Yet, because of incomplete and/or poorly annotated databases, the vast majority of the measured. MS/MS spectra remain unidentified or identified as proteins of unknown function (PUFs), which severely restricts the analysis of the datasets. Our open-science metaPUF framework aims to unravel the dark matter of proteins of unknown function within the human gut microbiome by re-analysing publicly available meta-omic datasets using recently developed tools to produce metaproteomic databases integrated with a newly generated human gut microbiome genome catalogue. This novel workflow will provide a comprehensive search space to improve protein identification as well as functional annotation of PUFs with the ultimate aim to prioritize expressed disease-related proteins for further functional elucidation. Finally, the developed approaches and resources will be integrated within pre-existing and well-established open-access frameworks and will provide a sustainable and solid foundation for future functional meta-omic works.
Collaborators: Rob Finn (EMBL-EBI, MGNIFY); Juan Antonio Vizcaino (EMBL-EBI, PRIDE).
Funding: FNR CORE INTER – metaPUF
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Through connecting genomic and metabolic information, metaproteomics is an essential approach for understanding how microbiomes function in space and time. We are funding members of the Metaproteomics Initiative, which goals are to promote dissemination of metaproteomics fundamentals, advancements, and applications through collaborative networking in microbiome research. The Initiative aims to be the central information hub and open meeting place where newcomers and experts interact to communicate, standardize, and accelerate experimental and bioinformatic methodologies in this field.
References:
- Van Den Bossche T, Arntzen MØ, Becher D, Benndorf D, Eijsink VGH, Henry C, Jagtap PD, Jehmlich N, Juste C, Kunath BJ, Mesuere B, Muth T, Pope PB, Seifert J, Tanca A, Uzzau S, Wilmes P, Hettich RL, Armengaud J. The Metaproteomics Initiative: a coordinated approach for propelling the functional characterization of microbiomes. Microbiome, 2021, 9:243, doi: 10.1186/s40168-021-01176-w
- Van Den Bossche T, Kunath BJ, Schallert K, Schäpe SS, Abraham PE, Armengaud J, Arntzen MØ, Bassignani A, Benndorf D, Fuchs S, Giannone RJ, Griffin TJ, Hagen LH, Halder R, Henry C, Hettich RL, Heyer R, Jagtap P, Jehmlich N, Jensen M, Juste C, Kleiner M, Langella O, Lehmann T, Leith E, May P, Mesuere B, Miotello G, Peters SL, Pible O, Queiros PT, Reichl U, Renard BY, Schiebenhoefer H, Sczyrba A, Tanca A, Trappe K, Trezzi JP, Uzzau S, Verschaffelt P, von Bergen M, Wilmes P, Wolf M, Martens L, Muth T. Critical Assessment of MetaProteome Investigation (CAMPI): a multi-laboratory comparison of established workflows. Nature Communications, 2021, 12:7305, doi: 10.1038/s41467-021-27542-8
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The ability to compare the expression of microbial functions between different conditions such as the human gut microbiome in a healthy and disease state is extremely important in biology in order to fully resolve and understand what exactly changes between conditions and generate proper clinical interpretations. Different quantification and statistical methods are available for microbiome analysis. However, metaproteomics comes with additional challenges and its quantitative analysis still suffers from many issues.
This project aims to generate high-quality datasets in order to assess and develop accurate quantitative methods for metaproteomics. By developing samples of known compositions, abundances and variations, we will know exactly what we are supposed to observe. Using that information, we will be able to test multiple quantification and analysis methods and find out which one most accurately capture protein abundance and variations between samples. Additionally, we will also be able to assess alternative recently developed approaches. Ultimately, we aim at providing the scientific community with a list of quantification and statistical methods that will allow them to fully characterize the functions of their biological samples and accurately compare them between different biologically relevant conditions.Collaborator: Manuel Kleiner (North Carolina State University)
Funding: FNR INTER Mobility (to Benoît Kunath)
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
In the natural world, individual cell populations are typically not found in isolation but are in direct contact with other cell types or organisms. Co-culture systems have been developed for addressing a number of fundamental biological questions relating to interactions between different cell populations but are typically limited in scope. This project is focussed on the development of a modular microfluidics-based co-culture device, termed HuMiX, which allows proximal co-culture of human epithelial cells and microbes. The HuMiX device mimics physiologically relevant spatial dimensions and establishes extracellular matrix conditions, which allow the establishment of stable growth conditions for both cell contingents. We have developed such devices from design to implementation via state-of-the-art micro fabrication techniques. The devices include integrated sensors for online monitoring of physicochemical parameters like concentration of oxygen and pH. As a proof-of-concept, we have recently demonstrated long-term co-culture of human epithelial cell lines (Caco-2) with LGG and/or B.caccae, which forms the basis for developing a microfluidics-based model of the entire human gastrointestinal tract.
Collaborator: Frederic Zenhausern (University of Arizona, Phoenix, USA)
Funding: FNR CORE programme grant ”HuMiX”; Proof-of-concept FNR programme grant “HuMix2.0”References:
- Fritz J.V., Desai M.S., Shah P., Schneider J.G., Wilmes P. From meta-omics to causality: experimental models for human microbiome research. Microbiome, 2013, 1(1):14. doi: 10.1186/2049-2618-1-14
- Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, Niegowska M, Estes M, Jäger C, Seguin-Devaux C, Zenhausern F, Wilmes P. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat Communications, 2016, 7:11535. doi: 10.1038/ncomms11535
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
A human individual’s microbiome consists of around 100 trillion cells, which represents at least ten times as many cells as human cells constitute the body. Beneficial effects of the presence of microbial communities on human physiology range from immune cell development and homeostasis, food digestion via the fermentation of non-digestible dietary components in the large intestine to balancing the host’s metabolism and promoting angiogenesis. Negative consequences for the host linked to the human microbiome include for example chronic inflammation and infection. Indeed, shifts in microbial community structure and function (dysbiosis) have been linked to numerous human diseases, including inflammatory bowel disease, diabetes mellitus, obesity, cardiovascular disease and cancer. The largest microbial reservoir of the human body is the gastrointestinal tract (GIT) and, thus, it is also the most studied and important from a biomedical perspective.
Therefore, it seems of great importance to understand and control the interplay between GIT microorganisms and human immune cells located in the gastrointestinal-associated lymphoid tissue (GALT), which constitutes the largest immune compartment in the human body. It is estimated that T cells associated with the small intestinal epithelium alone account for more than 60% of the total body lymphocytes. Currently, it is difficult to study the crosstalk between human immune cells and GI microorganisms in vivo in humans for obvious ethical reasons. Moreover, a systematic manipulation of variables to test the impact of a specific subtype of immune cells on the microbiota as well as the inflammatory effect of specific microbial species on human immune cells is not possible in vivo. In this respect, we plan to develop a microfluidics-based in vitro co-culture system, which allows the culture and modulation of different subtypes of primary immune cells in presence and absence of human gut microorganisms. Bacterial and human immune cells are separated by an epithelial cell monolayer to closely mimic the GIT.Collaborators: RIKEN Center for Integrative Medical Sciences, Laboratory for Gut Homeostasis, Team Leader: Kenya Honda
Funding: FNR: AFR – Development and establishment of a microfluidics-based in vitro culture model to study the impact of HIV infection on the gastrointestinal mucosal barrier – grant to Joëlle Fritz; FNR CORE programme grant, Proof-of-concept FNR programme grantReferences:
- Fritz JV, Desai MS, Shah P, Schneider JG, Wilmes P. From meta-omics to causality: experimental models for human microbiome research. Microbiome, 2013, 1(1):14. doi: 10.1186/2049-2618-1-14
- Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, Niegowska M, Estes M, Jäger C, Seguin-Devaux C, Zenhausern F, Wilmes P. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nature Communications, 2016, 7:11535. doi: 10.1038/ncomms11535.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The majority of the microorganisms constituting the human microbiome inhabit the gastrointestinal tract (GIT) where they play essential roles in governing human health. A variety of diseases including colorectal cancer (CRC) are associated with dysbiosis, a pathological imbalance in the intestinal microbiota. Apart from endogenous microbial consortia, diets supplemented with prebiotics, are thought to have a major effect on GIT microbiota and are inversely correlated with the risk of developing CRC. Furthermore, the use of probiotics including Lactobacillus rhamnosus GG has been found to exhibit anti-cancer effects. Here, we study the synergistic effects of probiotic bacterial strains, dietary components and colorectal adenocarcinoma enterocytes of the human GIT using the microfluidics-based GIT co-culture model (HuMiX). More specifically we are interested in the phenotypical characteristics of the enterocytes using specific cell invasion, migration and proliferation assays. We are also studying the gene expression changes in human enterocytes after different pre-and probiotic treatments within the HuMiX model.
A thorough mechanistic understanding of the interplay between dietary habits, bacterial metabolism and human physiology is required to understand the role of pre-and probiotics and apply them appropriately in CRC therapies and prevention. In this context, this project will likely result in recommendations for dietary and probiotic-based interventions to modulate the microbiota-host relationship in order to reduce the expression of pro- carcinogenic genes and reduce pro-inflammatory responses that have been found to play a pivotal role in CRC.
Collaborators: Serge Haan and Elisabeth Letellier (Life Sciences Research Unit, Luxembourg)
Funding: Internal Research Project of the University of Luxembourg “MiDiCa”; FNR AFR PhD grant to Kacy Greenhalgh and grant from the Luxembourg Personalised Medicine Consortium.Reference:
- Greenhalgh K, Ramiro-Garcia J, Heinken A, Ullmann P, Bintener T, Pacheco MP, Baginska J, Shah P, Frachet A, Halder R, Fritz JV, Sauter T, Thiele I, Haan S, Letellier E, Wilmes P. Integrated In Vitro and In Silico Modeling Delineates the Molecular Effects of a Synbiotic Regimen on Colorectal-Cancer-Derived Cells. Cell Reports, 2019, 27(5):1621-1632.e9. doi: 10.1016/j.celrep.2019.04.001.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The microbial communities that inhabit the human gastrointestinal tract play roles in both health and disease. Different species of gut bacteria (~10^3) possess wider metabolic capabilities, including those not encoded in the human genome e.g., degradation of complex carbohydrates present in our diets. Changes in the relative proportions of distinct functional groups of gut bacteria, termed dysbiosis, have been implicated in several intestinal disorders, including diabetes, inflammatory bowel disease (IBD) and colon cancer. Little is known about the mechanisms behind dysbiosis; most studies have taken case-control comparative analytical approaches to identify species with altered abundance during disease. However, most studies have not had the statistical power to infer causal relationships. Therefore, in vivo or in vitro experiments are necessary to examine the effects of different bacterial groups. To carry out such experiments, it is necessary to compile representative artificial gut microbial communities with which hypotheses can be tested. Therefore, we have designed a synthetic (simplified) human intestinal microbiota that contains species with diverse metabolic potentials. This artificial microbiota is being used to study the interaction of gut microbes with the host, using in vivo (gnotobiotic mice) and in vitro (microfluidics-based device; see the section on HuMiX) approaches.
Funding: FNR CORE – HuMiX
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
The human gut microbiome is a significant contributor to human health and disease. This emphasized by the notion that alterations in the gut microbiome are not solely related to gastrointestinal diseases, but also to neurological diseases. To study the exact interactions of the gut microbiome with the enteric nervous system (ENS), our personalized gut-on-chip in vitro model will enable proximal co-culture of ENS cells with microbial cells. Our research involves a two-pronged approach, i.e. (i) to further develop the human-microbial crosstalk (HuMiX) gut-on-chip model by introducing induced pluripotent stem cell (iPSC)-derived ENS cells inside the device. (ii) Upon successful introduction of the ENS cells, the resulting model, neuroHuMiX, will be used to investigate the gut microbiome-nervous system axis by leveraging multi-omic analyses which will allow for the identification of specific microbial and/or molecular markers. Furthermore, neuroHuMiX will serve as a means to enhance personalized microbiome studies, where different cell types from the same individual will be established within one device.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Over the past few years, several studies have shown that the gastrointestinal microbiome plays a key role in processes such as digestion and immunity and that imbalances in the gut microbial composition are linked to the development of idiopathic diseases. The gut microbiome also plays a key role in the processing of exogenous pharmaceutical compounds. More specifically, recent studies indicate that the intestinal microbiome can directly or indirectly affect drug metabolism by inactivating a drug or by reactivating a detoxified drug which consequently leads to the generation of toxic compounds and intestinal toxicity. Modeling the highly variable luminal gut environment and understanding how gut microbes can modulate drug availability or induce gut toxicity remains a challenge. Hence, there is a significant need to develop new platforms that enable the co-culture of human and microbial cells in a patient-specific manner. Using microfluidics-based technologies, specifically organ-on-chips (OoC), we aim to overcome current challenges in drug toxicity assessment assays as these technologies are designed to be more physiologically relevant than conventional in vitro and in vivo models. We have thus interconnected our microfluidics-based Human Microbial Crosstalk (HuMiX) model with a liver-on-chip model to create a stem-cell based multi-organ platform to predict the effect of the gut microbiome on pharmacokinetics in a personalized way.
Funding: MSCA-ITN “EUROoC”
Collaborator: Alexander Mosig (University Clinic Jena, Germany)
Reference:
- Lucchetti M, Kaminska M, Oluwasegun AK, Mosig AS, Wilmes P. Emulating the gut-liver axis: Dissecting the microbiome’s effect on drug metabolism using multiorgan-on-chip models. Current Opinion in Endocrine and Metabolic Research, 2021, 18:94-101. doi: 10.1016/j.coemr.2021.03.003.
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
Parkinson’s disease (PD) is the second most common neurodegenerative disease and presently remains incurable. Gastrointestinal symptoms precede the characteristic motor symptoms by years to decades and there is evidence supporting initiation of the disease by enteric inflammation and changes in microbial community composition and function. The central hypothesis involves so far unknown pathogenic agents entering through the gut, propagating via susceptible cell populations (dopaminergic neurons), and only in later stages affecting the brain. Changes in the gut microbiome of PD patients include increased levels of mucus-foraging bacteria, which may be linked to a compromised gut barrier, and decreased levels of fibre-degrading bacteria, leading to a decreased butyrate production and thus e.g. reduced energy availability for intestinal cells. Next to this, our research group has recently discovered elevated levels of a metabolite, 2-hydroxypyridine (2-HP), in the gut microbiome of patients with PD, which is potentially linked to PD development and progression. These observations suggest a microbiome-driven, multi-factorial mechanism for the pathogenesis of PD, including erosion of the gut barrier, production of elevated levels of 2-HP, as well as induction and sustainment of (neuro-) inflammatory pathways. This indicates the potential for microbiome-focused modulatory strategies for the treatment and prevention of PD. Such a strategy aims to induce a shift in the microbiome and metabolome of at-risk individuals or PD patients, and to decrease 2-HP production and its cytotoxic effects on the enteric nervous system.
In this project, we will develop a mechanism-based synbiotic formulation, a mixture of live biotherapeutic bacteria (probiotic) and a dietary formulation (prebiotic), to modulate the PD patient microbiome, with the threefold aim to decrease (i) gut permeability associated with PD, (ii) the production of the recently discovered 2-HP metabolite linked to PD development and progression, and (iii) intestinal and systemic (neuro-) inflammation. We aim for our synbiotic product to restore the metabolic profile and microbial community composition in PD patients and to have a protective effect on the intestinal mucus, epithelium and nervous system with direct impacts on clinical practice.
Funding: H2020 Widening Fellowship (to Charlotte de Rudder)
-
Project details (PDF):
-
Duration:
-
Funding source:
-
Researchers:
-
Partners:
-
Description:
As cell confluency is a crucial experimental parameter, in the context of using stem cells (organoids/iPSCs) in HuMiX but also for studying the effect of toxic compounds on barrier function, we worked on the integration of a non-invasive, online measurement method. Thus, together with Uppsala University, TEER electrodes have been conceived, designed, fabricated, and tested in HuMiX. We have successfully measured differentiation, barrier formation, as well as induced barrier disruption in real-time in HuMiX, thus demonstrating the feasability, and applicability of online TEER measurements in our gut-on-a-chip model.
Funding: MSCA-ITN “EUROoC”
Collaborator: Maria Tenje (Uppsala University)
-
Project details (PDF):