
 Faculté des Sciences, des Technologies et de Médecine
Faculté des Sciences, des Technologies et de Médecine Faculté de Droit, d’Économie et de Finance
Faculté de Droit, d’Économie et de Finance Faculté des Sciences Humaines, des Sciences de l’Éducation et des Sciences Sociales
Faculté des Sciences Humaines, des Sciences de l’Éducation et des Sciences Sociales Interdisciplinary Centre for Security, Reliability and Trust
Interdisciplinary Centre for Security, Reliability and Trust Luxembourg Centre for Systems Biomedicine
Luxembourg Centre for Systems Biomedicine Luxembourg Centre for Contemporary and Digital History
Luxembourg Centre for Contemporary and Digital History Luxembourg Centre for European Law
Luxembourg Centre for European Law Luxembourg Centre for Socio-Environmental Systems
Luxembourg Centre for Socio-Environmental Systems
DNA contains the blueprint for life, be it for mammals, plants or microorganisms. Thanks to current technologies, DNA can be extracted from virtually any environment, including mixed communities of microorganisms like a microbiome, and then sequenced. Sequencing produces what could be described as small copies of different parts of the blueprint. A software is then used to reconstruct the original blueprint, i.e., to which genes the different DNA sequences contained in a sample correspond. This is called a metagenomic reconstruction. It allows researchers to identify genes of interest and to understand the composition and functions of microbiomes. As technologies are fast evolving, scientists have now at their disposal different sequencing approaches and software to generate these metagenomic reconstructions. In an article recently published in Briefings in Bioinformatics, researchers of the Luxembourg Centre for Systems Biomedicine (LCSB) at the University of Luxembourg and from the Luxembourg Institute of Science and Technology (LIST) investigate how these different approaches impact the reconstruction and downstream analyses of microbiomes. They show that different sequencing technologies – long-read or short-read – as well as assembly strategies, meaning the choice of software used for the reconstruction of either long-read data, short-read data, or both in a hybrid approach, can lead to different conclusions. This is especially relevant as the use of emerging long-read sequencing technologies will increase in the future and discrepancies between approaches will need to be addressed. These findings pave the way for critical assessments of metagenomic reconstructions.
Constant evolution in sequencing technologies
Sequencing technologies have come a long way. The first generation – Fred Sanger’s chain-termination method – was first described in 1977 and became the main technology for sequencing over the next decades. A second generation was launched in 2005 with the arrival of next-generation sequencing (NGS), new techniques that lowered the costs and increased throughput. Despite these improvements, NGS technologies have a common limitation: the inability to sequence long stretches of DNA, hence they are also referred to as “short-read sequencing” (SRS) technologies.
Third-generation sequencing, also called long-read sequencing (LRS), is now emerging and tackles several of the shortcomings of the second generation. With LRS, read lengths can reach over 10 kilobase pairs and it is possible to resolve complex and repetitive loci in genomes. “LRS is considered to be the next frontier of genomics,” details Dr Susheel Bhanu Busi, joint-first author of the article along Dr Valentina Galata. “As it is especially relevant to study microbial populations, our research group at the LCSB was very interested in testing and assessing this new technology.”

Short-reads, long-reads and hybrid
Despite the promises of LRS, previous studies have shown that its accuracy remains lower compared to SRS and that error-correction steps are needed. Hybrid (HY) assembly methods using both SRS and LRS have been proposed to reduce the error rate while leveraging the increased read contiguity but the overall impact of the sequencing and assembly methods chosen to reconstruct the genes in a microbial community is not well understood.
Researchers from the Systems Ecology group and Bioinformatics Core at the LCSB and colleagues from the LIST evaluated short-read-only, long-read-only and hybrid assembly approaches in four different metagenomic samples of varying complexity: a mock community, a natural whey starter culture, a cow rumen sample and a novel dataset from a human faecal sample. “We wanted to understand how sample diversity and assembly approach are linked, and address the influence of sequencing technologies,” explains Dr Cédric Christian Laczny, senior author of the article.
The results of the comparisons carried out by the researchers reveal that short-read, long-read and hybrid approaches not only differ markedly in their overall performance, but also influence the prediction of genes and proteins. The discrepancies observed between the assemblies based on SRS, LRS and HY could impact phylogenetic and functional studies based on these reconstructions, especially when the differences concern functionally relevant genes such as antimicrobial resistance genes.
“Our results demonstrate that, irrespective of sample diversity, the sequencing approach and assembly strategy can have a significant impact on the characterisation of the microbiome’s functional potential,” says Dr Valentina Galata. “They also highlight the complementarity of long-read and short-read data: they can be combined to validate predictions and provide high-confidence reconstructions”
Complementary omics are key for functional analyses
Leveraging the System Ecology group’s experience in meta-omics studies of microbiomes, the researchers generated metatranscriptomic and metaproteomic data for one of the four studied samples. They used this data to assess how this complementary information on the microorganisms’ RNA, peptides and proteins, respectively, could be used to to resolve discrepancies between assembly approaches, for an improved study of the functional profile of the microbiome. Their results show that the incorporation of meta-omics data have a synergistic effect on protein verification and enable critical assessment of metagenome reconstructions. This is the first study to integrate meta-omics data in combination with long-read sequencing data and to demonstrate the added value that this combination brings to study microbiomes.
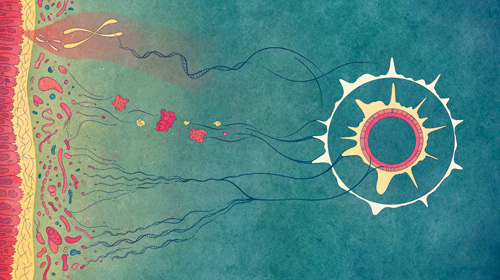
A combined approach to get a better overview of microbiomes
To tackle the discrepancies between sequencing and assembly strategies, the authors propose a reference-independent approach that will help to identify high-confidence genomic reconstructions. It makes use of the synergies between multiple sequencing technologies and allows a more comprehensive integration of meta-omics data.
“Past studies using long-read sequencing data have focussed on reconstructing the genomes of the most abundant organisms but have ignored large portions of the microbial community by doing so. We aim to get a better understanding of the entire community rather than a few select members,” concludes Dr Cédric C. Laczny. “This new approach will be highly relevant to achieve this as it will help us to get more reliable insights into the microbiomes’ compositions and functions. »
—
Reference: Valentina Galata, Susheel Bhanu Busi, Benoît Josef Kunath, Laura de Nies, Magdalena Calusinska, Rashi Halder, Patrick May, Paul Wilmes, Cédric Christian Laczny, Functional meta-omics provide critical insights into long- and short-read assemblies, Briefings in Bioinformatics, 2021. https://doi.org/10.1093/bib/bbab330
Illustration by Valentina Galata.