
 Faculté des Sciences, des Technologies et de Médecine
Faculté des Sciences, des Technologies et de Médecine Faculté de Droit, d’Économie et de Finance
Faculté de Droit, d’Économie et de Finance Faculté des Sciences Humaines, des Sciences de l’Éducation et des Sciences Sociales
Faculté des Sciences Humaines, des Sciences de l’Éducation et des Sciences Sociales Interdisciplinary Centre for Security, Reliability and Trust
Interdisciplinary Centre for Security, Reliability and Trust Luxembourg Centre for Systems Biomedicine
Luxembourg Centre for Systems Biomedicine Luxembourg Centre for Contemporary and Digital History
Luxembourg Centre for Contemporary and Digital History Luxembourg Centre for European Law
Luxembourg Centre for European Law Luxembourg Centre for Socio-Environmental Systems
Luxembourg Centre for Socio-Environmental Systems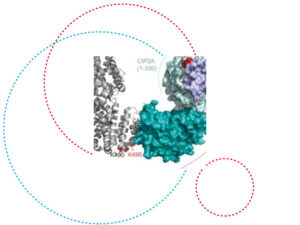
The Turku Bioscience Centre and the University of Luxembourg have recently published an article in the renowned multidisciplinary journal Nature Communications on the structural mechanism of how oncogenic CIP2A inhibits the B56a-complex of the tumour suppressor PP2A. This work is a result of an international collaboration across eight countries, three continents and over six years of research.
International collaboration
The research was conducted by the Cancer Cell Signaling group at the Turku Bioscience Centre (Turku, Finland), led by Prof. Jukka Westermarck, supported by the Cancer Cell Biology and Drug Discovery group of Prof. Daniel Abankwa at the University of Luxembourg. Postdoctoral researcher Dr. Karolina Pavic led the study at the University: “We are very much looking forward to what these findings may mean for the drug targeting efforts of the CIP2A-PP2A complex”, states Dr. Pavic.
Key roles of the tumour suppressor PP2A
Protein phosphorylation is a dynamic, reversible and tightly regulated post-translational modification, implicated in regulating protein functions in virtually all aspects of cellular life. Regulation of protein phosphorylation is achieved through opposing activities of protein kinases, which add a negatively charged phosphate group, and protein phosphatases. Their dysregulation contributes to the development of many human diseases, including cancer and neurological-disorders.
Protein phosphatase 2A (PP2A) is one of the major cellular phosphatases, which negatively regulates pro-survival and proliferation factors that are relevant for malignant transformation into cancer cells. The broad impact of PP2A’s activity is possible through its combinatorial structural organisation of several A, B and C-subunits, which gives rise to more than 80 different heterotrimeric holoenzymes. Different regulatory B-subunits specify substrates and thus mediate a multitude of activities of the PP2A trimers. Especially the B56 family of B-subunits has been linked to PP2A´s tumour suppressive function. B56a interacts with up to 100 cellular target proteins, including the protooncogene c-MYC, via binding to the LxxIxE-motif in the target protein. In cancer, PP2A is inactivated by several mechanisms, in particular by the over-expression of its endogenous inhibitor Cancerous Inhibitor of PP2A (CIP2A).
A detailed mechanistic understanding of how CIP2A inhibits the PP2A-B56a holoenzyme has remained elusive, despite its significance for drug development.
A complex puzzle solved by a multidisciplinary approach
The study used a multidisciplinary approach involving chemical cross-linking coupled with mass spectrometry, in vitro protein binding studies, peptide competition assays, phosphoproteomics, cell imaging, CRISPR-Cas9 mediated mutagenesis and in vivo studies in mice. It reports an elegant “hijack and mute” model, demonstrating that CIP2A competes with B56a substrate binding by blocking the LxxIxE-substrate motif binding pocket on B56a. In addition, CIP2A hijacks B56α and catalytic PP2Ac and takes up the role of a pseudo-scaffold in the newly formed CIP2A-B56α-PP2Ac trimer, thus muting substrate recognition by B56α. At the same time, CIP2A is protected from regulated degradation, which further explains the potency of CIP2A as a human oncoprotein.
Potential biomedical applications
The relevance of this mechanism for tumourigenesis was demonstrated in a triple negative breast cancer model, where a single point mutation in the B56α interaction domain of CIP2A abrogated tumour growth. Importantly, these proof-of-concept data validate the interface of the two proteins as a novel drug target site.
The detailed understanding of how the PP2A-B56α heterotrimer assembly and substrate recognition is regulated may therefore have broad biomedical implications for several diseases, including cancer and Alzheimer´s disease.
Publication: “Structural mechanism for inhibition of PP2A-B56α and oncogenicity by CIP2A”, Nature Communication, February 2023